A fenilcetonúria (PKU) é uma doença rara na qual o bebê nasce sem a habilidade de quebrar adequadamente um aminoácido chamado fenilalanina.
Causas:
A fenilcetonúria é hereditária, isto é, passa de pais para filhos. O pai e a mãe devem passar o gene defeituoso para que o bebê tenha essa doença. Isso é conhecido como traço recessivo autossômico.
Os bebês com PKU não possuem uma enzima chamada fenilalanina hidroxilase, necessária para quebrar um aminoácido essencial denominado fenilalanina. Essa substância é encontrada em alimentos que contêm proteínas.
Sem essa enzima, os níveis de fenilalanina e de duas substâncias associadas a ela crescem no organismo. Tais substâncias são prejudiciais ao sistema nervoso central e causam dano cerebral.
Diagnóstico:
A fenilcetonúria pode ser fácilmente detectada em um simples exame de sangue. Em todos os estados brasileiros, o exame de triagem para PKU (ou fenilcetonúria), chamado teste do pezinho, é exigido para todos os recém-nascidos como parte do painel de triagem. O teste normalmente é realizado por meio da retirada de algumas gotas de sangue do bebê antes da saída dele do hospital.
Se o teste inicial é positivo, exames de sangue e urina são feitos para confirmar o diagnóstico.

Sintomas:
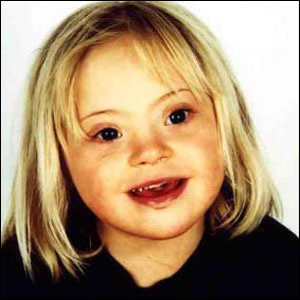
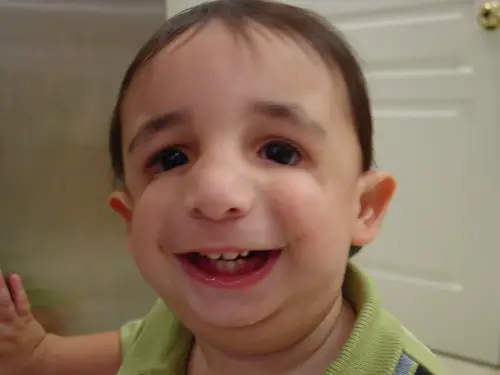
A fenilalanina atua na produção de melanina, o pigmento responsável pela cor da pele e do cabelo.
Portanto, bebês com essa doença geralmente posuem pele, cabelo e olhos mais claros do que seus irmãos que não sofrem dessa doença.
Outros sintomas podem incluir:
- Atraso mental e das habilidades sociais
- Tamanho da cabeça significantemente menor do normal
- Hiperatividade
- Movimentos incontroláveis de braços e pernas
- Retardo mental
- Convulsões
- Erupções cutâneas
- Tremores
- Posicionamento incomum das mãos
Se a doença não for tratada ou se os alimentos contendo fenilalanina não forem evitados, um odor "de rato" poderá ser sentido no hálito, na pele e na urina. Esse odor incomum deve-se ao aumento de substâncias de fenilalanina no organismo.
Tratamento:
A PKU é uma doença tratável. O tratamento consiste em uma dieta extremamente baixa em fenilalanina, especialmente durante o crescimento da criança. A dieta deve ser rigorosamente seguida. Isso demanda uma supervisão apurada de um nutricionista ou de um médico, além da cooperação dos pais e da criança. Aqueles que mantêm a dieta durante a vida adulta possuem saúde física e mental melhores. "Dieta para sempre" tornou-se uma bandeira levantada pela maioria dos especialistas. Isso é muito importante, principalmente antes da concepção e durante a gestação.